Tutorial
In this tutorial, we’ll analyze the Test Dataset which contains 13 multiplexed donors and ~20,982 droplets.
This tutorial will take you through the typical steps for demultiplexing data with Demuxafy. The process would be the same for non-multiplexed data for doublet detection but would be a different combination of softwares.
Select appropriate software combination
Run each selected software
Combine the results and call a final assignment for each droplet
Selecting Software Combination
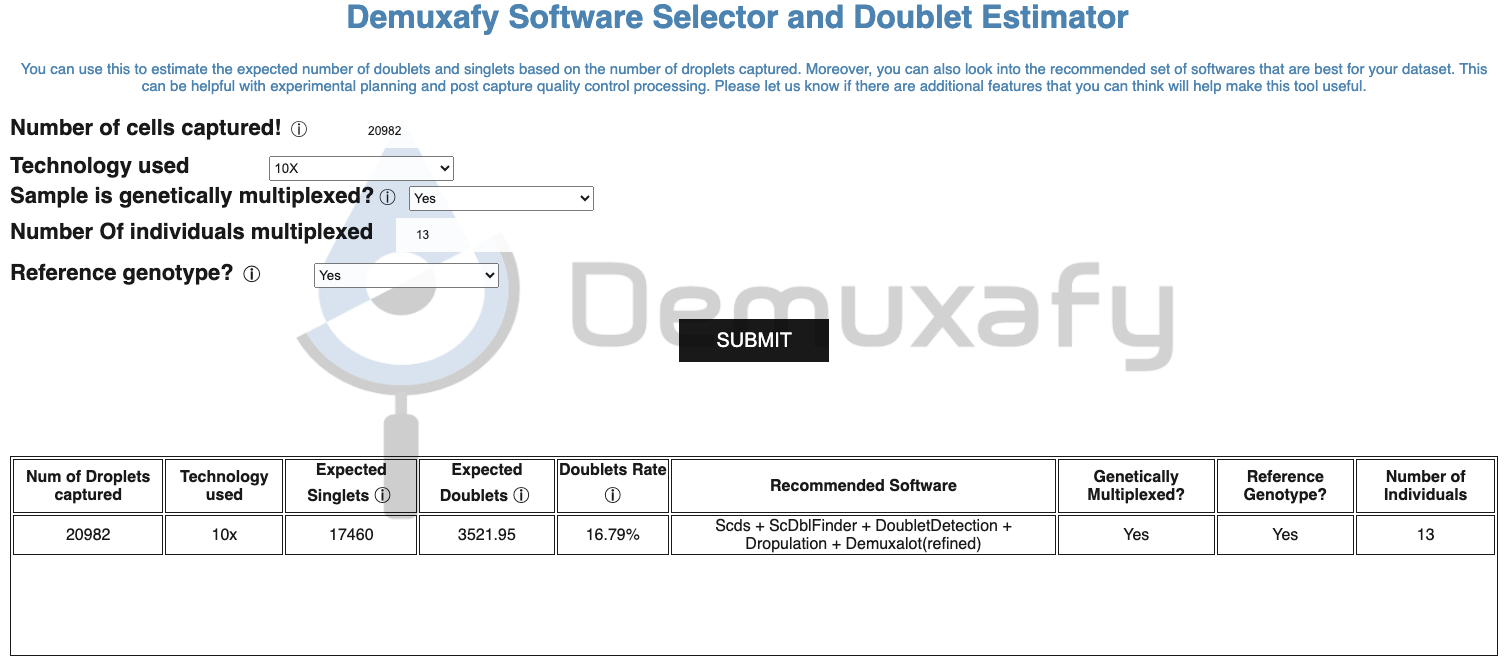
First, we’ll identify the softwares we should run using the Software Selection Tool. Start by entering the required information about the single-cell run into the Software Selection Tool. This sample is genetically multiplexed with reference SNP genotypes for each sample in the pool and 13 donors. When we enter that information and press “SUBMIT”, we can see the recommended method combination in the table below:
Note
If you want to compare different capture characteristics for experimental design, the recommendations and expected doublet numbers will be maintained in the table for easy comparison.
Per the recommendations we will run the following methods before combining them.
Demuxalot (refined)
Dropulation
DoubletDetection
ScDblFinder
Scds
Run Each Software
We will first run each software before combining them and calling final assignments for each droplet based on the annotations from each software. Please note that even though they are listed in sequence here, we run them in parallel since none of them are dependent on the other
Data
This is the data that you will need to have prepared to run each of the softwares and the softwares that use them:
Required
Reference SNP genotypes for each individual (
$VCF)Filtered for common SNPs (> 5% minor allele frequency) and SNPs overlapping genes
Barcode file (
$BARCODES)A file that has the barcodes annotated to contain cells in pool. One barcode per line, no header.
Bam file (
$BAM)Aligned single cell reads
Output directory (
$OUTDIR)A text file with the individual ids (
$INDS)File containing the individual ids (separated by line) as they appear in the vcf file
For example, this is the
individual filefor our example dataset
Here’s an example of our folder tree setup that will be used for this tutorial:
/path/to/data/
└── TestData4PipelineFull
├── donor_list.txt
├── samplesheet.txt
├── test_dataset
│ ├── outs
│ │ └── filtered_gene_bc_matrices
│ │ └── Homo_sapiens_GRCh38p10
│ │ ├── barcodes.tsv
│ │ ├── genes.tsv
│ │ └── matrix.mtx
│ ├── possorted_genome_bam.bam
│ └── possorted_genome_bam.bam.bai
└── test_dataset.vcf
We’ll start by setting up variables to different files and folders that we need for running each of the softwares
These are files provided as a test dataset available in the Data Preparation Documentation
Please replace /path/to with the full path to your data directory.
## Paths to files ##
VCF=/path/to/TestData4PipelineFull/test_dataset.vcf
COUNTS=/path/to/TestData4PipelineFull/test_dataset/outs/filtered_gene_bc_matrices/Homo_sapiens_GRCh38p10/
BARCODES=$COUNTS/barcodes.tsv
BAM=/path/to/TestData4PipelineFull/test_dataset/possorted_genome_bam.bam
INDS=/path/to/TestData4PipelineFull/donor_list.txt
GTF=/path/to/genes.gtf ## We do Not provide this - it should be the gtf file that you used to align your data. Otherwise you can download an appropriate gtf file from https://www.gencodegenes.org/human/
## Output directories ##
OUTDIR=/path/to/output
DEMUXALOT_OUTDIR=$OUTDIR/demuxalot
DROPULATION_OUTDIR=$OUTDIR/dropulation
DOUBLETDETECTION_OUTDIR=$OUTDIR/DoubletDetection
SCDBLFINDER_OUTDIR=$OUTDIR/scDblFinder
SCDS_OUTDIR=$OUTDIR/scds
Demuxalot (refined)
We’ll run Demuxalot with variant refinement:
⏱️ Expected Resource Usage
~2.5h using a total of 81Gb memory when using 32 threads
singularity exec Demuxafy.sif python Demuxalot.py \
-b $BARCODES \
-a $BAM \
-n $INDS \
-v $VCF \
-o $DEMUXALOT_OUTDIR \
-r True
HELP! It says my file/directory doesn’t exist!
If you receive an error indicating that a file or directory doesn’t exist but you are sure that it does, this is likely an issue arising from Singularity. This is easy to fix. The issue and solution are explained in detail in the Notes About Singularity Images
If Demuxalot is successful, you will have these new files in your $DEMUXALOT_OUTDIR:
/path/to/output/demuxalot
├── assignments_refined.tsv.gz
├── assignments.tsv.gz
├── likelihoods_refined.tsv.gz
├── likelihoods.tsv.gz
├── posterior_probabilities_refined.tsv.gz
└── posterior_probabilities.tsv.gz
Let’s check how many droplets were assigned as each donor and as doublets by Demuxalot with the demuxalot_summary.sh script:
singularity exec Demuxafy.sif bash demuxalot_summary.sh $DEMUXALOT_OUTDIR/assignments_refined.tsv.gz
which will return:
Classification |
Assignment N |
|---|---|
113_113 |
1344 |
349_350 |
1463 |
352_353 |
1619 |
39_39 |
1306 |
40_40 |
1082 |
41_41 |
1129 |
42_42 |
1437 |
43_43 |
1553 |
465_466 |
1091 |
596_597 |
1267 |
597_598 |
1523 |
632_633 |
872 |
633_634 |
961 |
660_661 |
1371 |
doublet |
2964 |
The estimated number of doublets (2,964) is slightly lower than the predicted number of doublets (3,522)
Dropulation
We’ll also run Dropulation:
Dropulation Assignment
First, Dropulation estimates the likelihood of each donor for each droplet
⏱️ Expected Resource Usage
~4h using a total of 3Gb memory when using 12 threads
Please note that the \ at the end of each line is purely for readability to put a separate parameter argument on each line.
singularity exec Demuxafy.sif AssignCellsToSamples --CELL_BC_FILE $BARCODES \
--INPUT_BAM $DROPULATION_OUTDIR/possorted_genome_bam_dropulation_tag.bam \
--OUTPUT $DROPULATION_OUTDIR/assignments.tsv.gz \
--VCF $VCF \
--SAMPLE_FILE $INDS \
--CELL_BARCODE_TAG 'CB' \
--MOLECULAR_BARCODE_TAG 'UB' \
--VCF_OUTPUT $DROPULATION_OUTDIR/assignment.vcf \
--MAX_ERROR_RATE 0.05
If the bam annotation is successful, you will have these new files in your $DROPULATION_OUTDIR:
/path/to/output/dropulation
├── assignments.tsv.gz
├── out_vcf.vcf
├── out_vcf.vcf.idx
└── possorted_genome_bam_dropulation_tag.bam
⏱️ Expected Resource Usage
~1.5h using a total of 5Gb memory when using 16 thread for the full Test Dataset which contains ~20,982 droplets of 13 multiplexed donors,
Next, we will identify the likelihoods of each droplet being a doublet.
Note
Please change the cell barcode and molecular barcode tags as necessary.
For 10x experiments processed with cellranger, this should be ‘CB’ for the CELL_BARCODE_TAG and ‘UB’ for the MOLECULAR_BARCODE_TAG
Please note that the \ at the end of each line is purely for readability to put a separate parameter argument on each line.
singularity exec Demuxafy.sif DetectDoublets --CELL_BC_FILE $BARCODES \
--INPUT_BAM $DROPULATION_OUTDIR/possorted_genome_bam_dropulation_tag.bam \
--OUTPUT $DROPULATION_OUTDIR/likelihoods.tsv.gz \
--VCF $VCF \
--CELL_BARCODE_TAG 'CB' \
--MOLECULAR_BARCODE_TAG 'UB' \
--SINGLE_DONOR_LIKELIHOOD_FILE $DROPULATION_OUTDIR/assignments.tsv.gz \
--SAMPLE_FILE $INDS \
--MAX_ERROR_RATE 0.05
Dropulation Doublet
Next, Dropulation estimates the likelihood of a doublet for each droplet
⏱️ Expected Resource Usage
~1.5h using a total of 5Gb memory when using 16 threads
Next, we will identify the likelihoods of each droplet being a doublet.
Note
Please change the cell barcode and molecular barcode tags as necessary.
For 10x experiments processed with cellranger, this should be ‘CB’ for the CELL_BARCODE_TAG and ‘UB’ for the MOLECULAR_BARCODE_TAG
Please note that the \ at the end of each line is purely for readability to put a separate parameter argument on each line.
singularity exec Demuxafy.sif DetectDoublets --CELL_BC_FILE $BARCODES \
--INPUT_BAM $DROPULATION_OUTDIR/possorted_genome_bam_dropulation_tag.bam \
--OUTPUT $DROPULATION_OUTDIR/likelihoods.tsv.gz \
--VCF $VCF \
--CELL_BARCODE_TAG 'CB' \
--MOLECULAR_BARCODE_TAG 'UB' \
--SINGLE_DONOR_LIKELIHOOD_FILE $DROPULATION_OUTDIR/assignments.tsv.gz \
--SAMPLE_FILE $INDS \
--MAX_ERROR_RATE 0.05
Dropulation Call
Finally, we will make final assignments for each droplet based on the doublet and assignment calls.
Please note that the \ at the end of each line is purely for readability to put a separate parameter argument on each line.
singularity exec Demuxafy.sif dropulation_call.R --assign $DROPULATION_OUTDIR/assignments.tsv.gz \
--doublet $DROPULATION_OUTDIR/likelihoods.tsv.gz \
--out $DROPULATION_OUTDIR/updated_assignments.tsv.gz
If the bam annotation is successful, you will have these new files in your $DROPULATION_OUTDIR:
/path/to/output/dropulation
├── assignments.tsv.gz
├── likelihoods.tsv.gz
├── out_vcf.vcf
├── out_vcf.vcf.idx
├── possorted_genome_bam_dropulation_tag.bam
└── updated_assignments.tsv.gz
Dropulation Summary
We can check the distribution of cells that were assigned to each donor and annotated as doublets with the Dropulation_summary.sh script:
singularity exec Demuxafy.sif bash Dropulation_summary.sh $DROPULATION_OUTDIR/updated_assignments.tsv.gz
which will return:
Classification
Assignment N
113_113
1327
349_350
1440
352_353
1562
39_39
1255
40_40
1082
41_41
1122
42_42
1365
43_43
1546
465_466
1084
596_597
1258
597_598
1515
632_633
815
633_634
892
660_661
1364
doublet
3355
The estimated number of doublets (3,355) is very close to the predicted number of doublets (3,522)
DoubletDetection
We will also run DoubletDetection:
singularity exec Demuxafy.sif DoubletDetection.py -m $COUNTS -o $DOUBLETDETECTION_OUTDIR
HELP! It says my file/directory doesn’t exist!
If you receive an error indicating that a file or directory doesn’t exist but you are sure that it does, this is likely an issue arising from Singularity. This is easy to fix. The issue and solution are explained in detail in the Notes About Singularity Images
This will return the following files:
/path/to/output/DoubletDetection
├── convergence_test.pdf
├── DoubletDetection_doublets_singlets.tsv
├── DoubletDetection_summary.tsv
└── threshold_test.pdf
Looking at the DoubletDetection_summary.tsv file, the number of doublets (2,594) is lower than the predicted number of doublets (3,522)
DoubletDetection_DropletType
Droplet N
doublet
2594
singlet
18388
ScDblFinder
We will also run ScDblFinder.
⏱️ Expected Resource Usage
~1min using a total of 3Gb memory when using 2 thread for the full Test Dataset which contains ~20,982 droplets of 13 multiplexed donors,
singularity exec Demuxafy.sif scDblFinder.R -o $SCDBLFINDER_OUTDIR -t $COUNTS
HELP! It says my file/directory doesn’t exist!
If you receive an error indicating that a file or directory doesn’t exist but you are sure that it does, this is likely an issue arising from Singularity. This is easy to fix. The issue and solution are explained in detail in the Notes About Singularity Images
After running the ScDblFinder you will have two files in the $SCDBLFINDER_OUTDIR:
/path/to/output/scDblFinder
├── scDblFinder_doublets_singlets.tsv
└── scDblFinder_doublet_summary.tsv
Looking at the scDblFinder_doublet_summary.tsv file, the number of doublets (3,323) is slightly lower than the predicted number of doublets (3,522)
Classification |
Droplet N |
|---|---|
doublet |
3323 |
singlet |
17659 |
Scds
Finally, we will also run Scds.
⏱️ Expected Resource Usage
~7min using a total of 10Gb memory when using 2 thread for the full Test Dataset which contains ~20,982 droplets of 13 multiplexed donors,
To run Scds with our wrapper script, simply execute the following in your shell:
singularity exec Demuxafy.sif scds.R -o $SCDS_OUTDIR -t $COUNTS
HELP! It says my file/directory doesn’t exist!
If you receive an error indicating that a file or directory doesn’t exist but you are sure that it does, this is likely an issue arising from Singularity. This is easy to fix. The issue and solution are explained in detail in the Notes About Singularity Images
After running the Scds with the wrapper script or manually you should have two files in the $SCDS_OUTDIR:
/path/to/output/scds
├── scds_doublets_singlets.tsv
└── scds_doublet_summary.tsv
Looking at the scds_doublet_summary.tsv file, the number of doublets (2,771) is lower than the predicted number of doublets (3,522)
Merging Results and Joint Software Calls
Now, we will combine the results from each of the softwares we’ve run (Demuxalot (refined), Dropulation, DoubletDetection, ScDblFinder, Scds) and annotate droplet cell types and donor assignments
singularity exec Demuxafy.sif Combine_Results.R \
-o $OUTDIR/combined_results.tsv \
--demuxalot $DEMUXALOT_OUTDIR \
--dropulation $DROPULATION_OUTDIR \
--solo $DOUBLETDETECTION_OUTDIR \
--scds $SCDBLFINDER_OUTDIR \
--scds $SCDS_OUTDIR \
--method "MajoritySinglet"
Results and Interpretation
After running the Combine_Results.R script, you will have have the following results
Here, we show the results for the above example that also provides combined calls with the “MajoritySinglet” calls.
/path/to/output/combined
├── combined_results_assignment_summary.tsv
├── combined_results_demultiplexing_summary.tsv
├── combined_results_droplet_type_summary.tsv
├── combined_results_Singlets_upset_donor_assignment.pdf
├── combined_results_Singlets_upset_droplet_type.pdf
├── combined_results_Singlets_upset.pdf
├── combined_results_summary.tsv
├── combined_results.tsv
└── combined_results_w_combined_assignments.tsv
Here’s a deeper look at the contents of each of some of those files:
First, we can look at the combined calls in the upset plot (combined_results_Singlets_upset.pdf):
This is an upset figure of the droplets which are colored by their final individual or doublet classification.
A filled circle indicates the that those droplets are classified as singlets by that software while empty circles indicate a doublet classification by that software
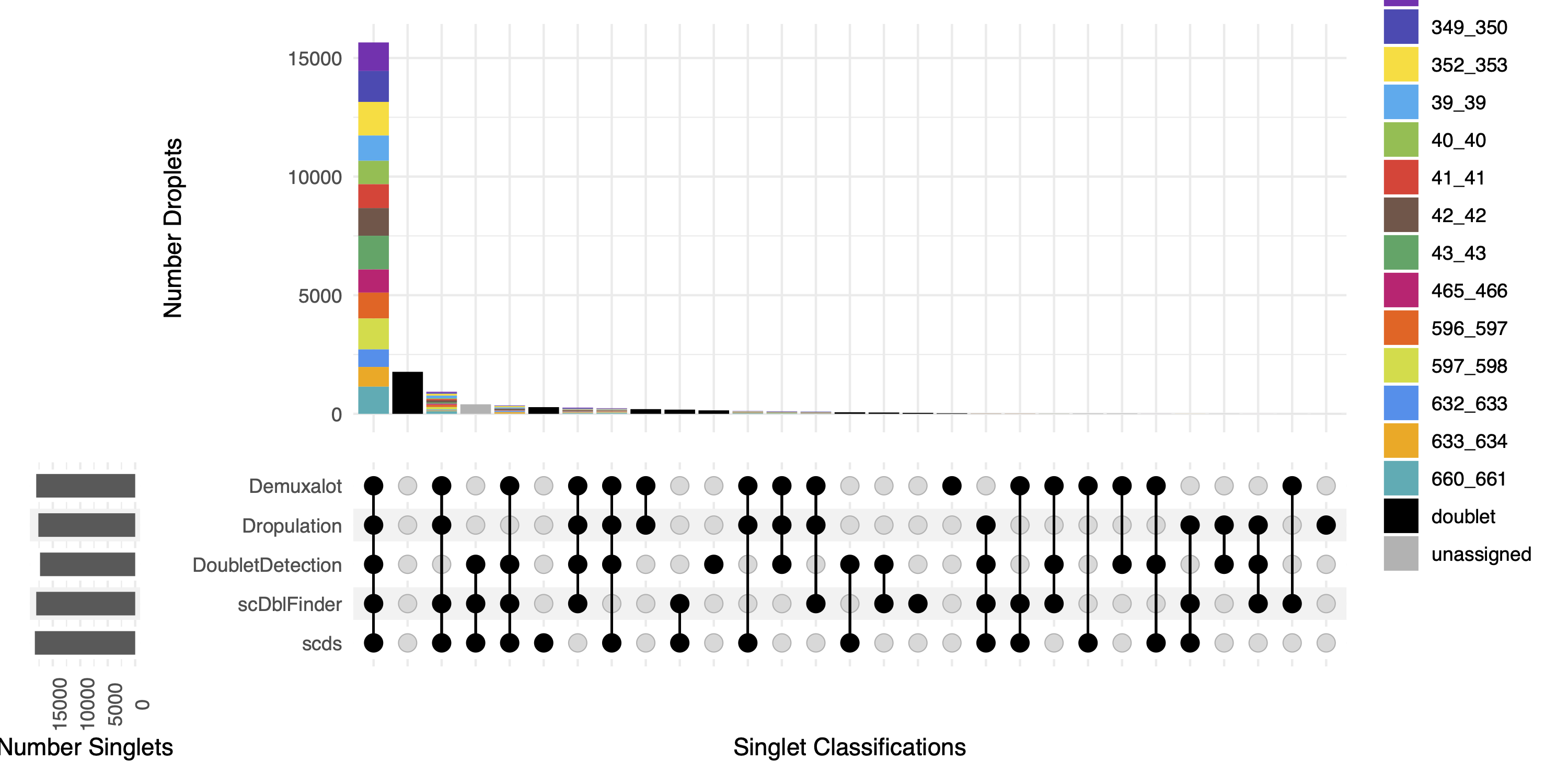
There are also summary files that provide the number of droplets annotated as a singlet or doublet by the combined softwares combined_results_droplet_type_summary.tsv:
Classification |
Droplet N |
|---|---|
doublet |
2771 |
singlet |
18211 |
and the number of droplets assigned to each donor and as doublets and unassigned by the combination of the softwares (combined_results_assignment_summary.tsv):
Classification |
Droplet N |
|---|---|
113_113 |
1333 |
349_350 |
1443 |
352_353 |
1607 |
39_39 |
1289 |
40_40 |
1072 |
41_41 |
1123 |
42_42 |
1409 |
43_43 |
1542 |
465_466 |
1084 |
596_597 |
1249 |
597_598 |
1493 |
632_633 |
859 |
633_634 |
953 |
660_661 |
1354 |
doublet |
2771 |
unassigned |
401 |
The combined_results.tsv file contains all the method calls + the combined finalized calls together in a single file that can be used as cell metadata for downstream analyses.
Barcode |
Demuxalot_Individual_Assignment |
Demuxalot_DropletType |
Dropulation_DropletType |
Dropulation_Individual_Assignment |
DoubletDetection_DropletType |
scDblFinder_DropletType |
scDblFinder_Score |
scds_score |
scds_DropletType |
MajoritySinglet_DropletType |
MajoritySinglet_Individual_Assignment |
|---|---|---|---|---|---|---|---|---|---|---|---|
AAACCTGAGATAGCAT-1 |
41_41 |
singlet |
singlet |
41_41 |
singlet |
singlet |
0.000161838892381638 |
0.11384647224872 |
singlet |
singlet |
41_41 |
AAACCTGAGCAGCGTA-1 |
465_466 |
singlet |
singlet |
465_466 |
singlet |
singlet |
0.038923978805542 |
0.503487172824797 |
singlet |
singlet |
465_466 |
AAACCTGAGCGATGAC-1 |
113_113 |
singlet |
singlet |
113_113 |
singlet |
singlet |
0.000687798717990518 |
0.0122651890679041 |
singlet |
singlet |
113_113 |
AAACCTGAGCGTAGTG-1 |
349_350 |
singlet |
singlet |
349_350 |
singlet |
singlet |
6.88672153046355e-05 |
0.099564348390602 |
singlet |
singlet |
349_350 |
AAACCTGAGGAGTTTA-1 |
632_633 |
singlet |
singlet |
632_633 |
singlet |
singlet |
0.000810008263215423 |
0.0887153542233592 |
singlet |
singlet |
632_633 |
AAACCTGAGGCTCATT-1 |
39_39 |
singlet |
singlet |
39_39 |
singlet |
singlet |
0.0342786461114883 |
0.0521116636059276 |
singlet |
singlet |
39_39 |
AAACCTGAGGGCACTA-1 |
465_466 |
singlet |
singlet |
465_466 |
singlet |
doublet |
0.962486505508423 |
0.600842973151551 |
singlet |
singlet |
465_466 |
AAACCTGAGTAATCCC-1 |
660_661 |
singlet |
singlet |
660_661 |
singlet |
singlet |
0.00426467135548592 |
0.431225466194795 |
singlet |
singlet |
660_661 |
… |
… |
… |
… |
… |
… |
… |
… |
… |
… |
… |
… |
You have completed the Tutorial! You can now apply the required methods to your dataset. Feel free to reach out if you have any questions, issues or recommendations with a Github issue.
Citation
If you used the Demuxafy platform for analysis, please reference our publication as well as DoubletDetection.